Introduction to my research in bioinformatics
8 May, 2017
Two and a half years ago I started my PhD in bioinformatics, a wide field of research closely related to biomedicine, which was totally unknown to me at that time. Biomedicine seeks the understanding (and treatment) of diseases at a molecular and biological scale, and bioinformatics adds the use of computational tools in this research. In this article I will make an introduction to my research, contextualizing it in the more general biomedical research field.
DNA, genes and proteins
All cells in our body (and any organism in general), contain huge molecules in the form of lengthy threads, which we call DNA. DNA is very important, because it acts like a big book of instructions for the development of living organisms.
 Source: bbc.com/education
Source: bbc.com/education
In the long thread which DNA is, there are pieces that are particularly important and are what we call genes. Genes are special because they contain some very specific information: the instructions to make proteins. Proteins, which can be imagined as tiny machines, are crucial to life because they perform all kinds of tasks: transporting components, accelerating chemical reactions, communicating signals, etc.
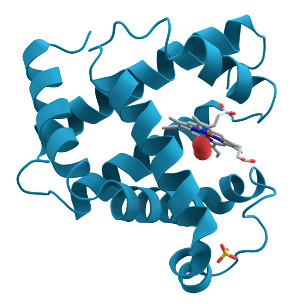 Example protein: myoglobin, which is responsible for transporting oxygen in muscle tissue.
Example protein: myoglobin, which is responsible for transporting oxygen in muscle tissue.
Source: wikipedia.org
Mutations
As we all know thanks to many TV series, the DNA of each person is different. But how different is it? In fact, the DNA of individuals is the same in 99.9% of the sequence, especially in the genes: we all need the same proteins to perform the same functions and therefore our genes are almost identical. When there are differences, they are usually in the form of single point mutations: specific changes in the DNA sequence. These mutations, when they occur on a gene, may also mean that the protein also has a mutation.
When we talk about mutations, we are not talking about abstract or unknown entities, as we have techniques nowadays that can find the mutations in the DNA of people. Some tools that in recent years have been taking more and more significance are the DNA sequencing machines, which are able to “read” a person's DNA. For example, they can be used to detect differences between cancer cells and healthy cells from the same patient to try to understand how the disease is developing in a particular case.
 DNA Sequencer: great strides over the past decade have meant that “reading” DNA is now be very affordable.
DNA Sequencer: great strides over the past decade have meant that “reading” DNA is now be very affordable.
Source: illumina.com
Mutations and diseases
Hence, are mutations in proteins good or bad? The answer is that it depends. Most mutations are harmless, we could even say that they are good because they don’t cause any harm, they allow the variety of living organisms and are key to evolution. However, some mutations are also the root cause of many diseases, for example by modifying the shape of a protein and preventing the fulfillment of its functions.
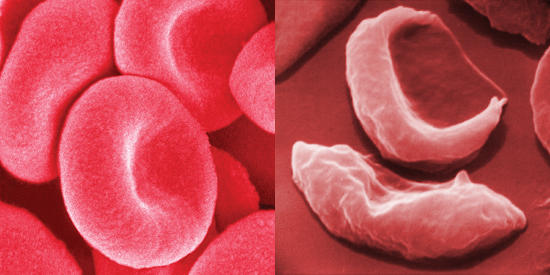 Healthy red blood cells and aberrant red blood cells (“banana”-shaped) due to a mutation in hemoglobin.
Healthy red blood cells and aberrant red blood cells (“banana”-shaped) due to a mutation in hemoglobin.
Source: britannica.com
Finding out whether a mutation is bad or not is difficult and expensive. It is part of the work that many researchers do around the world, trying to discover the molecular-scale implications of mutations in the development of various diseases. These findings are usually made public and put together, and so there are databases of mutations classified as benign or pathological. These databases, which in some cases contain more than 100,000 mutations, are still too small and contain only a small fraction of all the mutations that can be found in patients. Therefore, we need more tools to differentiate between benign and pathological mutations...
Can computers help in any way?
Pathology computer predictors
Computer science, and machine learning more specifically, provide us with tools that can help us classify mutations as either pathological or benign. Even though these methods will not be as reliable as the evidence obtained in the lab, they can be a great help for other researchers, pointing at promising leads and helping in the interpretation of large amounts of data. The first pathology predictors were developed 15 years ago, they are computer programs based on various methods that are able to predict (with an accuracy that does'nt usually exceed the 90%, but that can get close in some cases) the pathology of mutations.
In 2004, a team of researchers from Barcelona (including the advisors of my thesis), developed PMut, one of the first predictors that has been widely used since. When I started my PhD, I was commissioned to put the predictor up-to-date. Much has happened since 2004, the mutations databases are 10 times larger, and great advances in predictive methods have been made. The “simple” task of updating the PMut predictor has lead to research on how to improve predictors, deepen in the understanding of mutations and the strengths and weaknesses of these predictors.
Soon I will publish a blog article explaining my first contributions to this field and the specific lines of my research.